
EPJ D Highlight - Folding biomolecule model shows how form dictates function
- Details
- Published on 25 August 2017
New theoretical calculations of the conformation of very large biomolecules helps in understanding interactions with DNA strands as part of biological functions
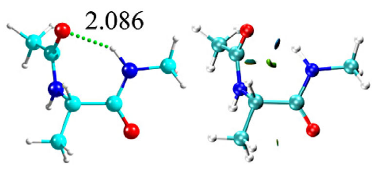
Proteins are fundamental macromolecules for life, with a diversity of functions, like acting as channels through cellular walls, catalysers, DNA benders, etc. When it comes to these functions, what matters is the layout of the secondary branches, made up of each protein’s amino acids, such as alanine, glutamine, arginine, phenylalanine and tyrosine. These are stabilised mainly by weak interactions-- such as hydrogen bonds, intramolecular interactions, and inter molecular dispersive forces, -- between the backbone and the lateral chain of their amino acids. In a new study published in EPJ D, Jorge González from the University of the Basque Country, in Leioa, Spain and colleagues have developed a theoretical method to calculate the most stable disposition that biomolecules try to adopt when they are together, or in close contact in cases where the bonding is weak. They also demonstrate that their model is consistent with our understanding of the same systems gained from experiments, such as spectroscopic analysis.
The sheer size of these molecules prevents correlation of the results of the configuration obtained for the isolated amino acids to the same amino acids constituting the secondary branches in the structure of proteins. Knowledge of the conformation adopted by the capped amino acids, authors believe, may be useful for the extrapolation of their properties to larger systems, like polypeptides or proteins.
The team first used molecular mechanics to identify the 3D conformation where the energy of the biomolecules would be the most stable. They then included quantum mechanics interactions in their model to better understand the structure and the vibrations of the various conformations together with their electron density. They then analysed the nature of intramolecular forces in each amino acid using bonding types like hydrogen bonds.
They found that the 15 most stable conformations studied had isolated capped amino acids whose structure resembles those found in proteins. However, they play a different role in the amino acids studied, depending on the character of their lateral chain.
Jorge González, Rodrigo Martínez, José A. Fernández and Judith Millan (2017),
Conformational landscape of isolated capped amino acids: on the nature of non-covalent interactions,
European Physical Journal D, DOI: 10.1140/epjd/e2017-80187-5


